
将 ScienceAI 设为星标
第一时间掌握
新鲜的 AI for Science 资讯



编辑 | ScienceAI
近些年来 AI for Science 在众多领域取得重大成功。其中,基于神经网络的量子变分蒙特卡洛方法 (NNVMC) 在量子化学领域展现出强大潜力,备受关注。
最近字节跳动研究部门 ByteDance Research 和北京大学团队在 NNVMC 框架中融入物理对称性,实现了量子激发态的高效精确求解。
该工作以《Spin-symmetry-enforced solution of the many-body Schr?dinger equation with a deep neural network》为题的论文已发表于国际顶级期刊 《Nature Computational Science》,相关代码已经开源。

代码地址:https://github.com/bytedance/jaqmc
此外,华东师范大学何晓老师及合作者在该期刊 News & Views 撰写了相关文章《Teaching spin symmetry while learning neural network wave functions》介绍了这一工作。
文章链接:https://www.nature.com/articles/s43588-024-00727-z
该工作由 ByteDance Research、北京大学物理学院陈基课题组和北京大学智能学院王立威课题组共同完成。
背景介绍
神经网络强大的表达能力让 NNVMC 方法在很多问题上的精度超越了传统量子化学方法。但以往工作未对自旋对称性这一基本物理性质加以约束。
一方面,NNVMC 算法本身只能在受限空间求解能量最低态,无法直接获取具有特定自旋对称性的量子态;另一方面,计算具有不同自旋对称性量子态近简并的体系时,容易得到有自旋污染的错误解。
为解决这些问题,作者创新地提出针对自旋对称性约束的惩罚项——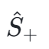 惩罚项,显著提高了求解特定自旋对称性基态 / 激发态的效率和精度。由于计算开销较低,该方案可用于较大体系,拓宽了 NNVMC 算法的应用范围。
惩罚项,显著提高了求解特定自旋对称性基态 / 激发态的效率和精度。由于计算开销较低,该方案可用于较大体系,拓宽了 NNVMC 算法的应用范围。
方法介绍
惩罚项
约束自旋对称性,较直接的做法是将神经网络波函数的自旋值作为惩罚项(即 惩罚项),直接添加到损失函数中训练优化,然而这种做法在计算上开销巨大因而被局限在较小的体系。
惩罚项),直接添加到损失函数中训练优化,然而这种做法在计算上开销巨大因而被局限在较小的体系。
作者提出一种新的等效的损失函数——损失函数,在相同精度条件下其计算开销相较之前低了一个量级。得益于直接限制了自旋对称性,优化过程中不同自旋对称性的量子态被「过滤」掉,因而实现了精度的提升,同时能够直接计算具有特定自旋对称性的量子态,提高了计算效率。

计算结果
基态训练的提升
作者对垂直构型的乙烯进行基态计算,通过加入惩罚项后,不仅能得到更低的能量,还能获得正确的自旋对称性。相比之下,NNVMC 直接计算的结果是基态(自旋单重态)与第一激发态(自旋三重态)的叠加态。
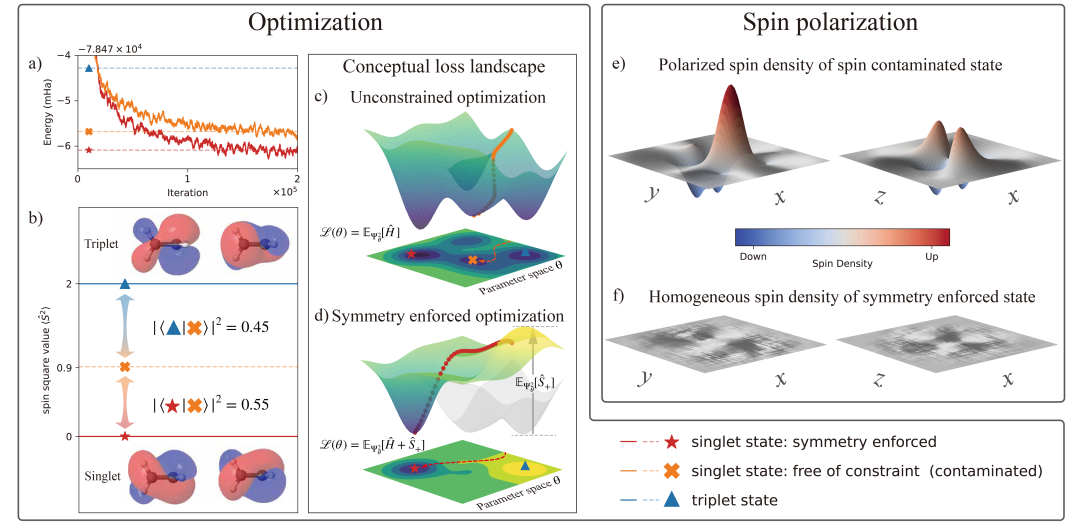
激发态训练的提升
作者针对两类体系做了高激发态计算,分别是原子光谱以及有机分子乙烯和甲醛,并将实验结果和当前 NNVMC 领域中具有代表性的激发态计算方法进行对比[1, 2]。
通过引入惩罚项,不同自旋对称性的量子态可以分别独立训练,从而减少了在激发态求解过程中需要保持正交量子态的数目,在精度以及效率上都有显著提升。

双自由基体系
由于其独特的电子结构,双自由基体系中自旋三重态与自旋单重态之间的能隙计算一直是传统量子化学领域的一大挑战。
由于两个量子态具有不同的自旋对称性,作者针对该类问题进行了计算并与被公认针对该类问题有效的求解方法辅助场量子蒙特卡洛也即 AFQMC 方法进行了对比[3, 4]。如图所示,通过惩罚项能够得到更高精度的结果。
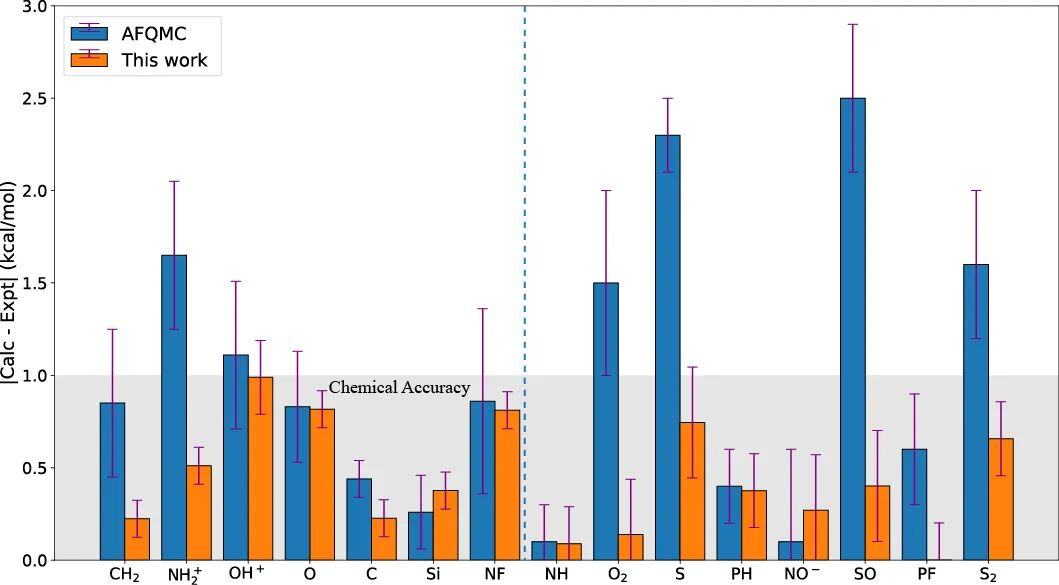
总结
为了拓展 NNVMC 方法的应用范围,字节跳动研究部门 ByteDance Research 和北京大学联合提出了惩罚项。相较于之前 NNVMC 的工作,结合该惩罚项后能够更快、更准地求解具有特定电子结构的量子化学问题。该工作在相关领域得到了关注,有望在解决复杂量子化学问题中发挥积极作用。
参考文献
[1] David Pfau et al., Accurate computation of quantum excited states with neural networks. Science 385, eadn0137 (2024). DOI: 10.1126/science.adn0137
[2] Szabó PB et al., An improved penalty-based excited-state variational Monte Carlo approach with deep-learning ansatzes. Journal of Chemical Theory and Computation. 2024 Aug 30;20(18):7922-35.
[3] Shee J, Arthur EJ, Zhang S, Reichman DR, Friesner RA. Singlet–triplet energy gaps of organic biradicals and polyacenes with auxiliary-field quantum Monte Carlo. Journal of chemical theory and computation. 2019 Aug 5;15(9):4924-32.
[4] Lee J, Malone FD, Morales MA. Utilizing essential symmetry breaking in auxiliary-field quantum Monte Carlo: Application to the spin gaps of the C36 fullerene and an iron porphyrin model complex. Journal of chemical theory and computation. 2020 Apr 13;16(5):3019-27.
人工智能 × [ 生物 神经科学 数学 物理 化学 材料 ]
「ScienceAI」关注人工智能与其他前沿技术及基础科学的交叉研究与融合发展。
欢迎关注标星,并点击右下角点赞和在看。
点击阅读原文,加入专业从业者社区,以获得更多交流合作机会及服务。