
转自公众号:知识图谱科技
http://mp.weixin.qq.com/s?__biz=MzI3ODE5Mzc1Ng==&mid=2247493366&idx=1&sn=a862c0a02fb5d31dcaf30b079a461398


摘要
这篇论文介绍了一个基于LLM(大型语言模型)的知识综合和科学推理框架,名为BioLunar,用于支持生物医学发现,特别是在肿瘤学中的生物标志物发现。
这篇论文展示了基于LLM的科学工作流框架BioLunar,支持专门的基因分析,特别是在肿瘤学和基因富集方面。该框架通过整合外部数据库、外部工具和上下文化的语言推理链,简化并自动化了复杂的分析过程。研究表明,LLM可以在生物医学环境中实现高度专业化的端到端分析工作流,支持异构证据的整合和结论的综合,同时记录和链接到数据源。
这篇论文为生物医学发现提供了一种新的方法,展示了LLM在复杂科学推理中的潜力。
核心速览
研究背景
-
研究问题:这篇文章要解决的问题是如何利用大型语言模型(LLMs)来支持生物医学发现,特别是肿瘤学中的生物标志物发现。
-
研究难点:该问题的研究难点包括:协调控制体内/体外干预、复杂的多步骤数据分析管道以及在前人证据的背景下解释结果;整合和融合分布式证据空间和工具;以及在低代码环境中实现复杂的科学工作流。
-
相关工作:该问题的研究相关工作包括Galaxy、Snakemake和Nextflow等科学工作流管理系统,这些系统主要用于系统化生物信息学分析过程,但在整合LLMs和外部数据库方面存在不足。
研究方法
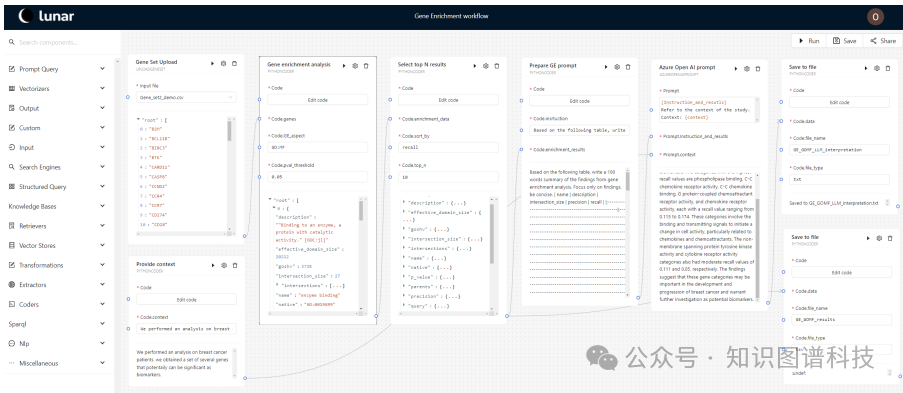
这篇论文提出了BioLunar,一个基于Lu-nar框架的工具,用于支持生物学分析,特别是肿瘤学中的分子级证据富集。具体来说,
-
集成LLMs:BioLunar通过标准化的API组件创建基于LLMs的生物医学科学工作流。LLMs可以整合和推理分布式证据空间和工具,降低跨多个结构化数据库和文本基础(如PubMed)访问和推理的障碍。
-
模块化设计:平台采用模块化设计,包含可重用的数据访问和数据分析组件,以及低代码用户界面,使所有编程水平的研究人员都能构建LLM启用的科学工作流。
-
自然语言推理(NLI):BioLunar利用自然语言推理(NLI)组件通过LLMs整合和调和证据空间,解释结果,生成全面的基因集输入摘要。
-
子工作流组件:子工作流组件允许在另一个工作流中重用现有工作流,简化了更复杂工作流的组合,避免了定义相同任务的重复步骤。
实验设计
-
数据收集:BioLunar集成了多种与精准肿瘤学相关的知识库,包括CIViC4、OncoKB5、Gene Ontology、Hu-man Protein Atlas、COSMIC、KEGG、Reactome和WikiPathways等。
-
实验设计:实验设计包括一个示例生物医学工作流,旨在整合生物信息学管道结果的证据并推断结论。具体步骤包括查询专家知识库、使用NLI组件整合和调和证据空间、生成全面摘要。
-
样本选择:实验中使用了两种不同场景的样本,每种场景产生了一组不同的基因,分别来自不同的生物信息学分析。
-
参数配置:用户可以通过拖放功能从功能分组类别中选择和排列组件,或使用Python或R脚本创建自定义组件。
结果与分析
-
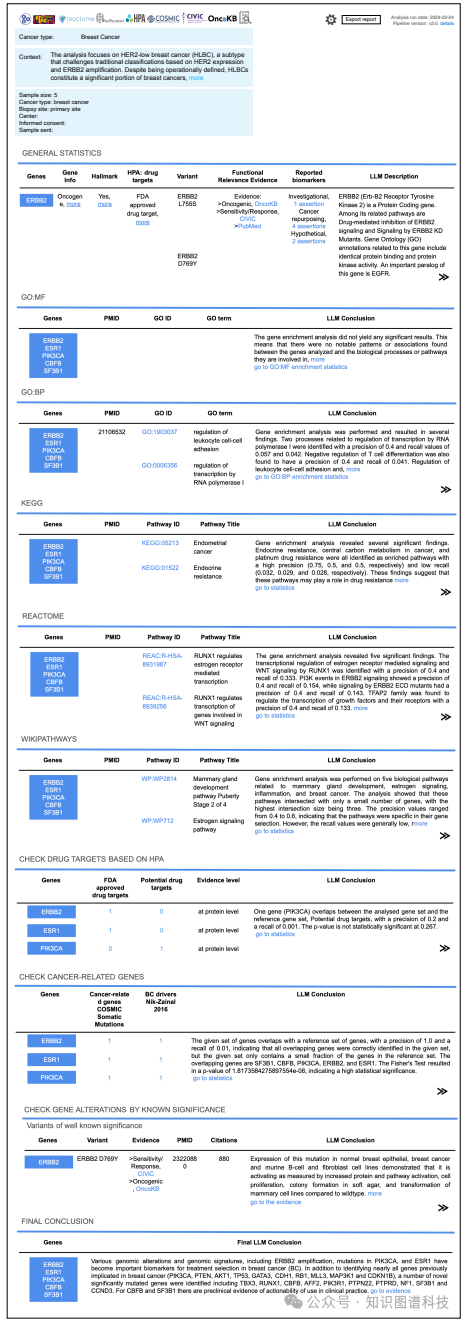
场景1:在探索HER2低表达乳腺癌的独特分子特征时,报告显示了识别到的基因组改变和基因组特征,包括ERBB2扩增、PIK3CA和ESR1突变,这些是乳腺癌治疗中的重要生物标志物。对于其余两个基因,发现了新的显著突变基因,并在临床实践中有预实验证据表明其具有可操作性。
-
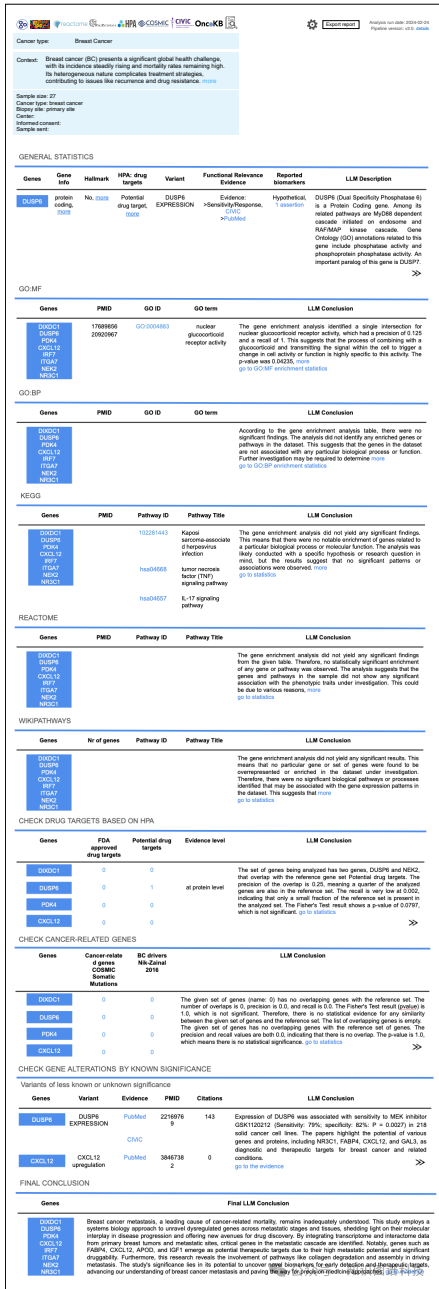
场景2:在发现可能导致更准确乳腺癌诊断的新基因时,报告显示了DIXDC1、DUSP6、PDK4、CXCL12、IRF7、ITGA7、NEK2、NR3C1等基因。这些基因中没有致癌基因,其中两个是潜在的药物靶点,一个是FDA批准的药物靶点。根据KEGG富集分析,这些基因主要通过几个信号通路富集,包括肿瘤坏死因子(TNF)信号通路。
总体结论
这篇论文展示了基于LLMs的科学工作流在支持肿瘤学和基因富集分析中的应用。BioLunar通过集成外部数据库、外部工具和上下文化LLM解释链,简化了复杂分析过程的自动化和整合。研究表明,LLMs可以在可重复的方式下启用复杂的端到端高度专业化的分析工作流,支持异构证据的整合、结论的综合,并在综合输出报告中记录和链接数据源。该工作流基于低代码范式,使领域专家无论其编程技能如何,都能构建和利用生成式AI方法启用的科学工作流。
论文评价
优点与创新
-
集成Large Language Models (LLMs): BioLunar利用LLMs来促进跨分布式证据空间的复杂科学推理,增强了在异构数据源之间进行协调和推理的能力。
-
模块化设计: 平台采用模块化设计,重用数据访问和数据分析组件,并提供了低代码用户界面,使所有编程水平的研究人员都能构建LLM启用的科学工作流程。
-
自动科学发现: 通过自动化科学发现和从异构证据中进行推理,BioLunar展示了LLMs、专门数据库和生物医学工具集成的潜力,以支持专家级别的知识综合和发现。
-
用户界面: 用户界面通过拖放功能组件,简化了工作流程的构建,支持单独执行、编辑或配置调整。
-
知识库集成: 当前框架集成了多种与精准肿瘤学相关的知识库,如CIViC、OncoKB、Gene Ontology等,提供了丰富的背景知识和专业分析工具。
-
子工作流程组件: 支持在另一个工作流程中重用现有工作流程,简化了更复杂工作流程的构建,避免了重复定义相同任务。
-
报告生成: 生成的报告包含研究背景、分析细节、日期和软件版本,并配有超链接以便于导航。
-
可扩展性: 系统架构支持轻松扩展,添加分支和组件而不影响现有工作流程,便于根据需求进行定制。
不足与反思
-
外部LLM-based APIs的使用: 当前演示使用了外部LLM-based APIs,但可以适应开源LLM模型。
-
LLM推断的定量评估: LLM推断需要关键的定量评估支持,目前的工作流程是由人类监督的假设生成过程,没有直接的临床应用。
关键问题及回答
问题1:BioLunar在处理异构数据源时是如何实现证据整合和推理的?
BioLunar通过集成Large Language Models (LLMs)来实现跨分布式证据空间和工具的复杂科学推理。具体来说,BioLunar利用LLMs进行自然语言推理(Natural Language Inference, NLI),以整合和协调来自不同知识库(如CIViC、OncoKB、Gene Ontology等)的证据。LLMs能够理解和解释这些知识库中的文本数据,并将其与观察到的基因或变异的显著性相关联。通过这种方式,BioLunar能够生成一个综合的摘要,解释整个基因集输入的结果,并考虑用户提供的生物分析上下文。
问题2:BioLunar的用户界面是如何设计的,它如何支持用户构建和运行LLM启用的科学工作流?
BioLunar的用户界面设计得非常直观,用户可以通过拖放功能从功能分组类别中选择和排列组件。组件被组织成不同的功能模块,如“Prompt Query”(包含NLI组件)、“Knowledge Bases”(知识库组件)、“Extractors”(从压缩档案中检索文件或从PDF文件中提取文本和表格的组件)和“Coders”(允许用户使用Python或R脚本创建自定义组件的组件)。用户无需编程知识即可创建工作流,因为组件是预定义的,只需提供数据输入或参数设置。对于需要编写自定义代码的用户,BioLunar提供了“Python Coder”和“R Coder”组件,并允许用户将这些自定义组件保存并在“Custom”组标签中访问。
问题3:BioLunar在实验中展示了哪些具体的应用场景,这些场景展示了BioLunar的哪些功能?
BioLunar在实验中展示了两个具体的应用场景:
-
场景1:用户探索HER2低表达乳腺癌的独特分子特征,以确定其是否构成乳腺癌类型中的一个独特类别。BioLunar识别出ERBB2扩增、PIK3CA和ESR1突变等重要生物标志物,并对剩余两个基因发现了新的显著突变基因,这些基因在临床实践中具有潜在的可操作性。
-
场景2:用户发现可能导致更准确乳腺癌诊断的新基因。BioLunar发现了一组基因(如DIXDC1、DUSP6、PDK4等),这些基因中没有致癌基因,其中两个是潜在的药物靶点,一个是FDA批准的药物靶点。KEGG富集分析显示,这些基因主要通过几个信号通路(如肿瘤坏死因子TNF信号通路)进行富集。BioLunar还通过PubMed搜索组件,查找描述了这些基因变体的最新出版物,进一步验证了其作为生物标志物的潜力。